| Autore |
Discussione |
|
|
car86
Nuovo Arrivato
Citt�: foggia

12 Messaggi |
 Inserito il - 16 maggio 2010 : 13:29:58 Inserito il - 16 maggio 2010 : 13:29:58




|
ciao a tutti.. avrei bisogno di alcuni consigli per una pcr che non mi viene.. e sono 1po sotto pressione..allora sto utilizzando una nuova taq, la fast start taq.. e ho impostato come condizioni di pcr:
95�C per 10 '
95�C per 30''
57�C per 30''
72�C per 30''
numero cicli 35
72�C per 10' per elongazione finale.
il risultato � una leggera bandina in corrispondenza del peso del mio gene, ma vedo anche delle forti bande in corrispondenza dei primers.. inoltre l'ultima volta ho trovato contaminazione nel bianco!
come posso risolvere??!!!
un'altra domanda: per verificare l'integrazione dell'mRNA devo vedere le 3bande facendolo correre sul gel d'agarosio?
spero in un vostro aiuto.. ve ne sarei grata!!
car86
|
|
|
|
|
domi84
Moderatore


Citt�: Glasgow
1724 Messaggi |
Inserito il - 16 maggio 2010 : 15:34:13
|
Ciao Car86 e Benvenuta sul forum!
Per la contaminazione nel bianco devi stare pi� attenta a cambiare le punte quando cambi reagenti. Se ti ri-succede avrai contaminato qualcosa.
Per come ho capito la banda all'altezza dei primer dovrebbero essere i primer, quindi forse ne metti troppi. Ricontrolla il protocollo. Se non ricordo male devi mettere 100-150 ng di primer...
La parte del ciclo
Citazione:
72�C per 30''
� dipendente dalla lunghezza del gene: 1 min per Kbp. Il tuo amplificato � lungo circa 500 bp?
--------
Citazione:
un'altra domanda: per verificare l'integrazione dell'mRNA devo vedere le 3bande facendolo correre sul gel d'agarosio?
spero in un vostro aiuto.. ve ne sarei grata!!
eh? integrazione dell'mRNA? quale mRNA? In cosa?
Scusa, ma se non ci spieghi bene non ti possiamo rispondere
 |
Il mio blog: http://domi84.blogspot.com/
Le foto che ho scattato... |
 |
|
|
car86
Nuovo Arrivato
Citt�: foggia
12 Messaggi |
Inserito il - 16 maggio 2010 : 15:48:58
|
ciao! allora..nella miscela di reazione io metto: 3microL di ciascun primer, 3microL di buffer di reazione, 3microL di magnesio, 3 microL di dNTPs e 0.5 microL di taq..e uso 2microL di cDNA..
ho un amplificato di 208 bp. la prima volta che ho usato la faststart taq non ho avuto segnale, poi modificando le condizioni ho avuto il risultato di prima, con un eccesso di primer.. domani deve assolutamente uscirmi qualcosa altrimenti la prof sclera!
per la contaminazione, sono abbastanza attenta a cambiare puntale.. ho il dubbio che mi si sia contaminato qualche reagente, non so come per�.. baster� cambiare i reagenti per la mix?
per quanto riguarda l'RNA, hai ragione, mi sono espressa male.. intendevo dire valutare che l'RNA sia integro, dopo l'estrazione, e non degradato..
grazie mille!!
|
|
|
|
domi84
Moderatore
Citt�: Glasgow
1724 Messaggi |
Inserito il - 16 maggio 2010 : 18:09:39
|
Tieni conto che dire "ho messo 3microL" stai dicendo il volume che hai messo, non la quantit�. Se questi sono troppo concentrati puoi mettere una quantit� eccessiva di primer. Quindi ricontrolla la quantit� di primer che metti, in nanogrammi o microMoli.
Citazione:
il risultato � una leggera bandina in corrispondenza del peso del mio gene, ma vedo anche delle forti bande in corrispondenza dei primers.. inoltre l'ultima volta ho trovato contaminazione nel bianco!
Puoi vedere cosa � contaminato facendo diverse PCR di controllo in cui ognuna manca un componente:
1 2 3 4 5
dNTP si si si si no
Primer1 si si si no si
Primer2 si si si no si
Taq si si no si si
Buffer si no si si si
Stampo no si si si si
Questo dovrebbe permetterti di identificare chi � contaminato da cosa...
Fai capire alla tua prof che le PCR si mettono a punto, per capire cosa non va...e non che ricompaiono magicamente dopo il week-end :D
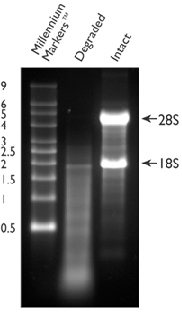
Per quanto riguarda l'RNA, s�, lo devi correre su un gel di Agarosio e MOPS e vedrai 3 bande dovute alla 28S, 18S e 5S...ma qualche volta la 5S non si vede.
L'importante � che la 28S sia il doppio di intensit� della 18S
 |
Il mio blog: http://domi84.blogspot.com/
Le foto che ho scattato... |
|
|
|
car86
Nuovo Arrivato
Citt�: foggia
12 Messaggi |
Inserito il - 16 maggio 2010 : 18:43:17
|
parto da una quantit� di primers di 5picomol..
nella pcr che ho fatto l'ultima volta mi � stato detto di mettere un numero di cicli diverso per campione: ne erano 3, e ho impostato 35, 40 e 45 cicli..togliendo i campioni uno alla volta e poi rimettendoli per l'elongazione finale, ma per me questo procedimento non � esatto.. e il segnale era sempre pi� debole all'aumentare dei cicli..potrebbe essere questo il problema?
grazie ancora, sei stato molto chiaro!
|
|
|
|
car86
Nuovo Arrivato
Citt�: foggia
12 Messaggi |
Inserito il - 18 maggio 2010 : 11:18:45
|
ciao!!
avrei una domanda da fare: � possibile vedere se il cDNA � integro o degradato facendolo correre sul gel d'agarosio? perch� potrebbe essere degradato, o magari non � venuta la retrotrascrizione, e quindi � il motivo per cui la pcr non viene..
grazie!
|
|
|
| |
Discussione |
|


